# Read subsetted data from online file - make sure there are no spaces
gtex = read_tsv('https://tinyurl.com/342rhdc2')
# Check number of rows
nrow(gtex)[1] 389922summarize() boils down the data frame according to the conditions it gets. In this case, it creates a data frame with a single column called blood_avg that contains the mean of the Blood columnmutate(), the name on the left of the = is something you make up that you would like the new column to be named.mutate() transforms columns into new columns of the same length, but summarize() collapses down the data frame into a single row

# A tibble: 4,999 × 2
Gene max_blood
<chr> <dbl>
1 A2ML1 2.08
2 A3GALT2 2.77
3 A4GALT 2.78
4 AAMDC NA
5 AANAT 1.71
6 AAR2 2.52
7 AARSD1 1.89
8 AB019441.29 2.31
9 ABC7-42389800N19.1 1.98
10 ABCA5 2.3
# ℹ 4,989 more rowsgroup_by() is a helper function that “groups” the data according to the unique values in the column(s) it gets passed.summarize() works the same as before, except now it returns as many rows as there are groups in the data
Before continuing, run this code to reformat your data and store it as a new data frame gtex_tidy (we’ll see how to do this later today):
Have a look at the dataframe you created. Use it to recreate this plot:

The max_expression variable in the x-axis of the plot indicates the maximum expression across all samples (in whatever grouping we are looking at).
It’s helpful to think backwards from the output you want. First outline the ggplot code that would generate the given plot. What does the dataset need to look like that is going into ggplot in order to get the plot shown here? How can we get from gtex_tidy to that data?
# A tibble: 3 × 8
tissue `2011` `2012` `2013` `2014` `2015` `2016` `2017`
<chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
1 Adipose Tissue 56 107 243 206 84 134 2
2 Adrenal Gland 28 41 84 65 20 31 0
3 Bladder 2 18 0 1 0 0 0Tidy data is easy to work with.
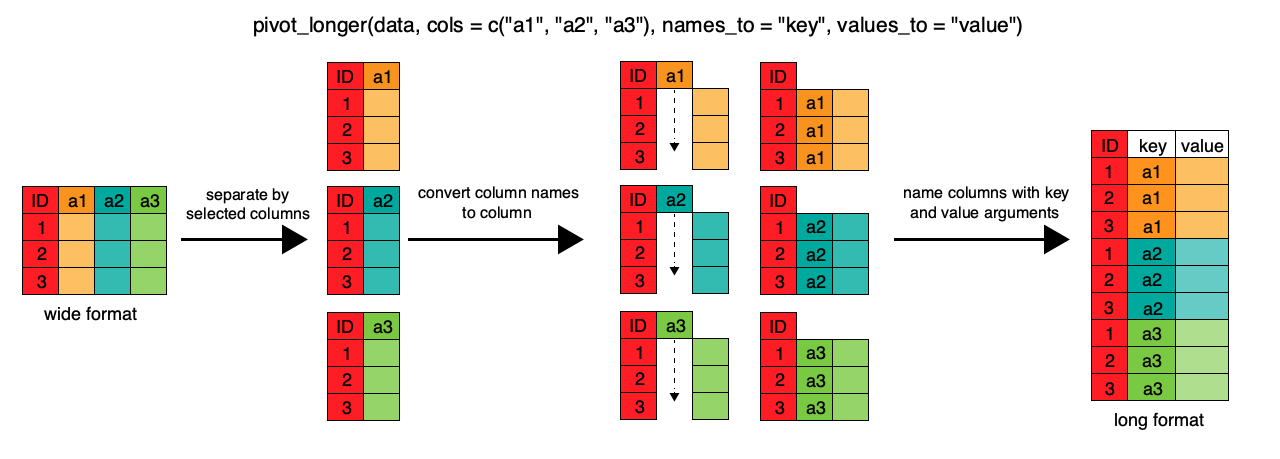
tidyr::pivot_longer() is the function you will most often want to use to tidy your datatidyr::pivot_longer() is the function you will most often want to use to tidy your data# A tibble: 2 × 3
tissue year count
<chr> <chr> <dbl>
1 Adipose Tissue 2011 56
2 Adipose Tissue 2012 107Use the GTEX data to reproduce the following plot:
The individuals and genes of interest are c('GTEX-11GSP', 'GTEX-11DXZ') and c('A2ML1', 'A3GALT2', 'A4GALT'), respectively.
Think backwards: what do the data need to look like to make this plot? How do we pare down and reformat gtex so that it looks like what we need?
subject data are referenced in the sample data, and the batches referenced in the sample data are in the batch data. The sample ids from the sample data are used for accessing expression data.subject connects to sample via a single variable, subject_id.sample connects to batch through the batch_id variable.